
At the Genetics Core we love a challenge, and we aren’t scared off by protocols that demand a fair investment of time. So, when we were approached to prepare some CAGE libraries for a research group we’ve collaborated with for many years we jumped at the chance. Perhaps a little too quickly!
So, what is CAGE and why would you want to do it?
Cap Analysis of Gene Expression (CAGE) was developed to provide high-throughput measurements of gene expression coupled with accurate mapping of transcriptional start sites and their promoters4. During transcription segments of template DNA are copied into complementary stretches of RNA1. Broadly speaking messenger RNA (mRNA) is the link between the information encoded by individual genes and the proteins that determine an organism’s fate2 – a description of the processes of transcription through to translation is shown in figure 15 below. However, most genes have multiple transcription starting sites (TSSs) making it difficult to identify the promoter regions that control the expression of the various transcripts2. The nucleotide at the 5’ end of some primary transcripts such as pre-cursor mRNA is modified or ‘capped’ as part of a highly regulated process vital for subsequent translation of mature mRNA during protein synthesis3.
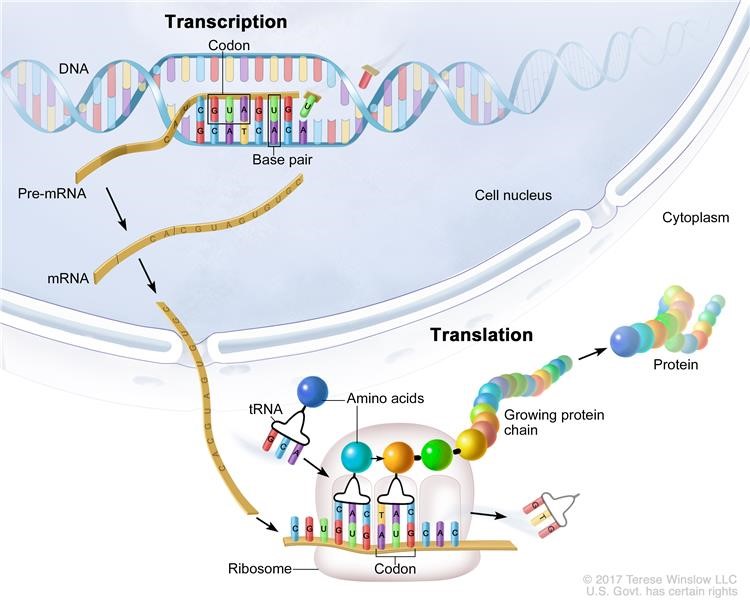
CAGE uses random or oligo dT primers to generate cDNA from an extracted total RNA sample. Full-length cDNAs are then captured using the ‘cap-trapping’ method and short sequence tags (20-27nt long) are generated from the 5’ ends that allow TSSs to be mapped, and transcript expression to be measured by tag frequency2. This includes all mRNAs and a large fraction of the non-coding RNAs in a sample. Figure 2 below taken from the protocol published by Takahashi et al (2012)4 shows the (deceptively long!) CAGE library preparation workflow.
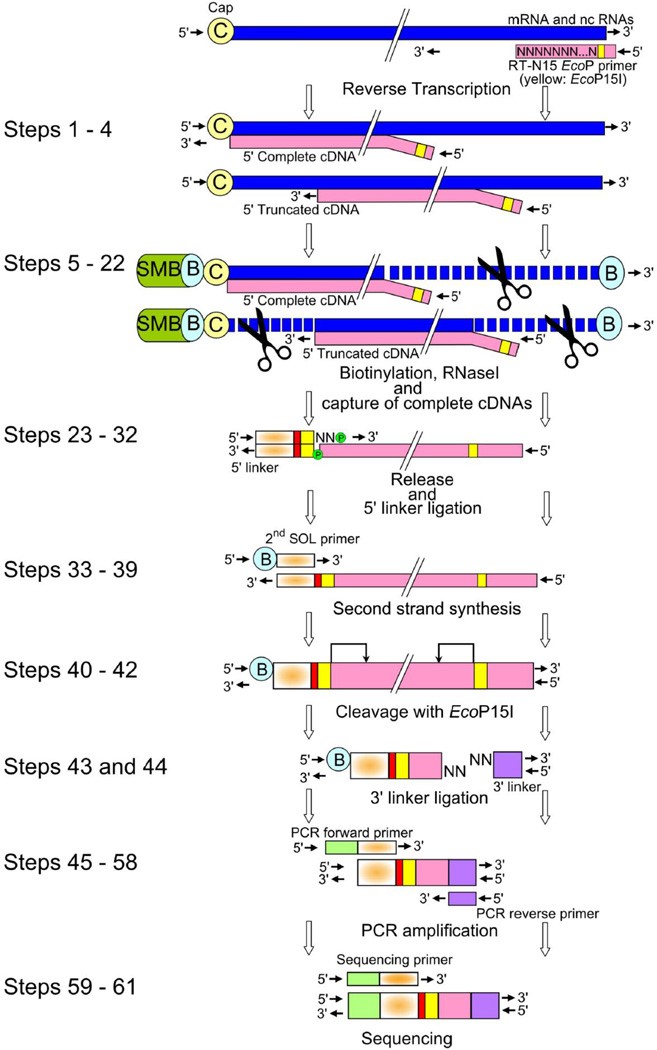
Our collaborators had been working with this technology for a while, but it was new to us so a training day – well, a training week actually – was arranged. The entire CAGE protocol takes 4 days when starting from total RNA and is designed to generate pools of 8 indexed CAGE libraries for running on a single lane of an Illumina HiSeq 2500. More about that later.
Training was provided by Sara Clohisey and Ivet Gazova, and took place in the Genetics Core at the Edinburgh Clinical Research Facility. We were joined by Jonathan Gillan – a post-graduate student working on Cystic Fibrosis research at the IGMM – and Claire O’Brien from the Australian National University Medical School in Canberra, who works on IBD research. The plan was for Sara and Ivet to each prep a set of libraries while we watched and took notes, then providing the libraries passed quality control checks to sequence on our lab’s own Illumina sequencer, the NextSeq 550. The week went well, as did the QC checks so I was keen to know what information the data might hold. We loaded our instrument and waited for the data…
…which was underwhelming to say the least. We pride ourselves here on returning high quality data to researchers but alas the run provided anything but! Very little sequencing had occurred, and that was made up entirely of the PhiX control library that Illumina suggest you add to sequencing runs – both to aid troubleshooting and to improve data quality when library diversity may be low. I sanity checked my paperwork and walked through the steps I’d taken. Had I set up the sequencer properly? Yes. Had I added the custom primer required for the libraries?? Yes. Had I loaded the library in the sequencer??? Yes.
Queue some back and forth between Illumina Technical Support and the lab, trying to work out what happened. They asked the same questions I’d already answered. Eventually, we worked out that the barcode and sequencing primers used in the protocol were all designed for single read flow cells, and were incompatible with the paired read flow cells available for the NextSeq. In fact, the only Illumina platform that these libraries would be compatible with is the HiSeq 2500 and this has now been obsolesced…sequencing facilities across the world have already decommissioned many of the instruments that were in operation! For an explanation of Illumina sequencing technology check out this video.
Since the training I’ve successfully prepared over 120 CAGE libraries for our collaborators that we’ve had to send to a facility based at the University of Liverpool who are still hanging on to their HiSeq 2500 for dear life. However, time is ticking and our own lab still needs a viable sequencing solution to be able to take advantage of the power of CAGE sequencing. So, I set about re-designing the oligos outlined in the Takahashi et al. protocol to make them compatible with paired-read flow cells. Besides compatibility with our NextSeq, this would allow CAGE libraries to be sequenced on the whole family of Illumina sequencers with the potential for massive demultiplexing to take advantage of higher throughput and the subsequent reduction in sequencing cost per sample.
Firstly I looked at the forward and reverse PCR primers used to introduce the sequence necessary for binding to the flow cell oligos – these are known as the P5 and P7 adapters respectively. These sequences are shorter for single-read flow cells, so I extended these to include the full sequences for paired-read flow cells.
Next I had to make sure that the linkers that would be ligated to our cap-trapped cDNAs would provide the full P5 and P7 sequences for the PCR primers to bind to, so I extended these. The original protocol provides sequences for 15 barcoded 5’ linkers, but we had been restricting this to a set of 8 that another lab had optimized to find were the most efficient at ligating to the cap-trapped cDNAs. I decided to try these same 8 linker barcodes so went ahead and ordered more.
Finally I redesigned the custom sequencing primer needed to run the libraries on our NextSeq to reflect the changes in the PCR primers and barcoded linkers. After a quick sanity check with technical support at Illumina (who never officially support custom libraries!), I ordered my oligos from IDT and waited for them to arrive.
Library Preparation of the paired-read flow cell compatible libraries was then a fairly straightforward case of substituting the new oligos for the old. I first prepared a set of 8 using RNA samples that had already been sequenced on the HiSeq 2500 using the original library preparation protocol, to give us a data set for a straight comparison that would allow us to assess the success of the ‘new’ protocol.
QC checks showed that the new oligos worked! Well, at least that I could make library that we could consider running on our NextSeq. This was enough for me, so we loaded the sequencer and started a run. I spiked in a high percentage of PhiX control library to ensure library diversity was high enough for the quality filters applied by the software on the instrument when resolving clusters, and to let us be sure whether the run had gone to plan. This is crucial for all Illumina sequencing instruments, but especially for those like the NextSeq that rely on two colours for sequencing, and there is more than one way around the issue; more on this in a future post.
The initial metrics looked good, so I generated FASTQ files and sent them on to our collaborator’s bioinformatician – Mazdak Salavati of the Roslin Institute – for analysis.
The results weren’t the resounding success I’d hoped for, but the upshot of the analysis was that it worked! Sort of. We’d spiked in a tremendous amount of PhiX (50%) so lost a lot of reads that way, but I had expected that. We were able to find some reads with the correct structure that could be assigned to all 8 barcode linkers I’d used in the library prep, although the proportion of this was low and seemed to be variable between the different barcodes. A conference call with Emily and Mazdak was fruitful, yielding a few avenues for further optimization of the protocol.
The use of a 3nt barcode sequence in the 5’ linkers meant that Mazdak had to use a very high stringency for demultiplexing the data, and this will likely have led to a fair number of reads being discarded for each sample. Increasing the barcode in each linker to 6nt would allow for a single base mismatch between the called base and the expected index, if the edit distance between the barcodes was higher.
The high level of variability in the number of reads assigned to each barcode might be a consequence of the new oligos having different ligation efficiency from the original set, so it might be worth looking at trying a larger set of 5’ linker barcodes to enable the identification of the best set. Ultimately we would want to run 16 samples together on our NextSeq (rather than 8) so it makes sense to increase the pool we can draw from.
Reducing the amount of PhiX control library spiked in to the runs would also help, as it would allow us to maximise the coverage of our CAGE libraries.
In order to build on our initial success we have applied for funding from the Scottish Universities Life Sciences Alliance (SULSA) to help with development costs. While we wait to hear about our application, it’s back to the drawing board to redesign the barcode linkers. I do this with a feeling of mixed success, but it’s good to have a proof-of-principle behind us and a plan moving forward. I’ll keep you updated on the road ahead.
References
- https://en.wikipedia.org/wiki/Transcription_(biology)
- http://fantom.gsc.riken.jp/protocols/basic.html
- https://en.wikipedia.org/wiki/Five-prime_cap
- Takahashi et al. (2012) 5′ end–centered expression profiling using cap-analysis gene expression and next-generation sequencing. Nature Protocols 7, 542-561
- https://www.cancer.gov/publications/dictionaries/cancer-terms/def/transcription
Richard Clark is a Research Technician in the Genetics Core and will sequence (almost) anything you throw at him. When he’s not at work he plays his guitar(s) or with weapons. Find him on twitter @SeqNinja
